Understand sources of stochastic gene expression
|
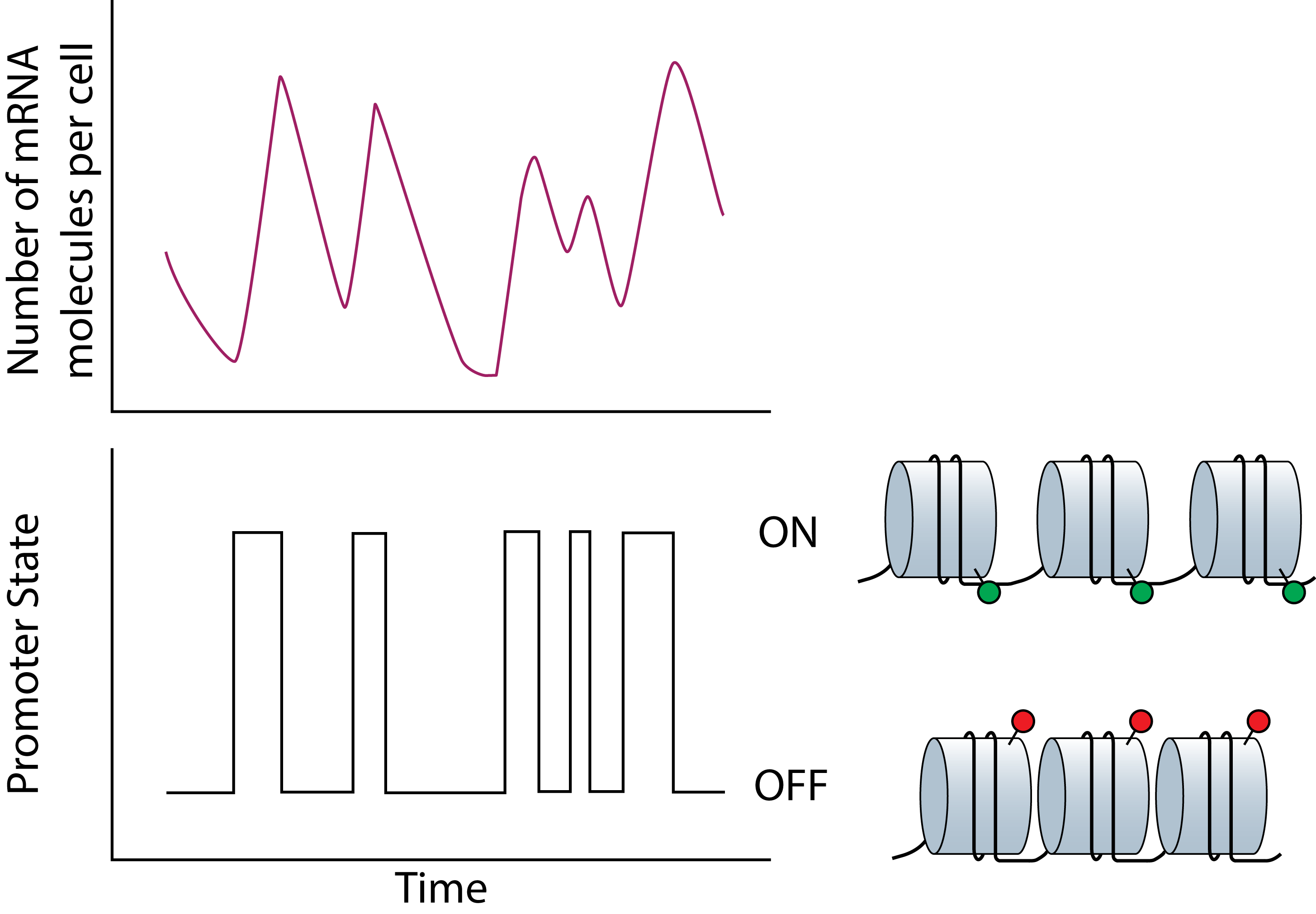
Over the last two decades, work from several research groups have shown that genes are expressed stochastically. This noise in gene expression coupled with complex biological networks have been shown to drive divergent cellular phenotypes. For example, stochastic gene expression has been shown to be one mechanism by which viruses evade host defenses to how mammalian stem cells adopt different cell fates. While stochastic gene expression and its consequences on cellular phenotypes have been studied extensively, the upstream genetic and epigenetic features regulating cell-to-cell heterogeneity in gene expression remains challenging to study and poorly understand. Through a combination of stochastic modeling and development of new experimental methods, we study how different epigenetic factors regulate gene expression noise with the ultimate goal of using these findings to modulate cellular functions and behavior. |
 |
Develop novel single-cell sequencing methods
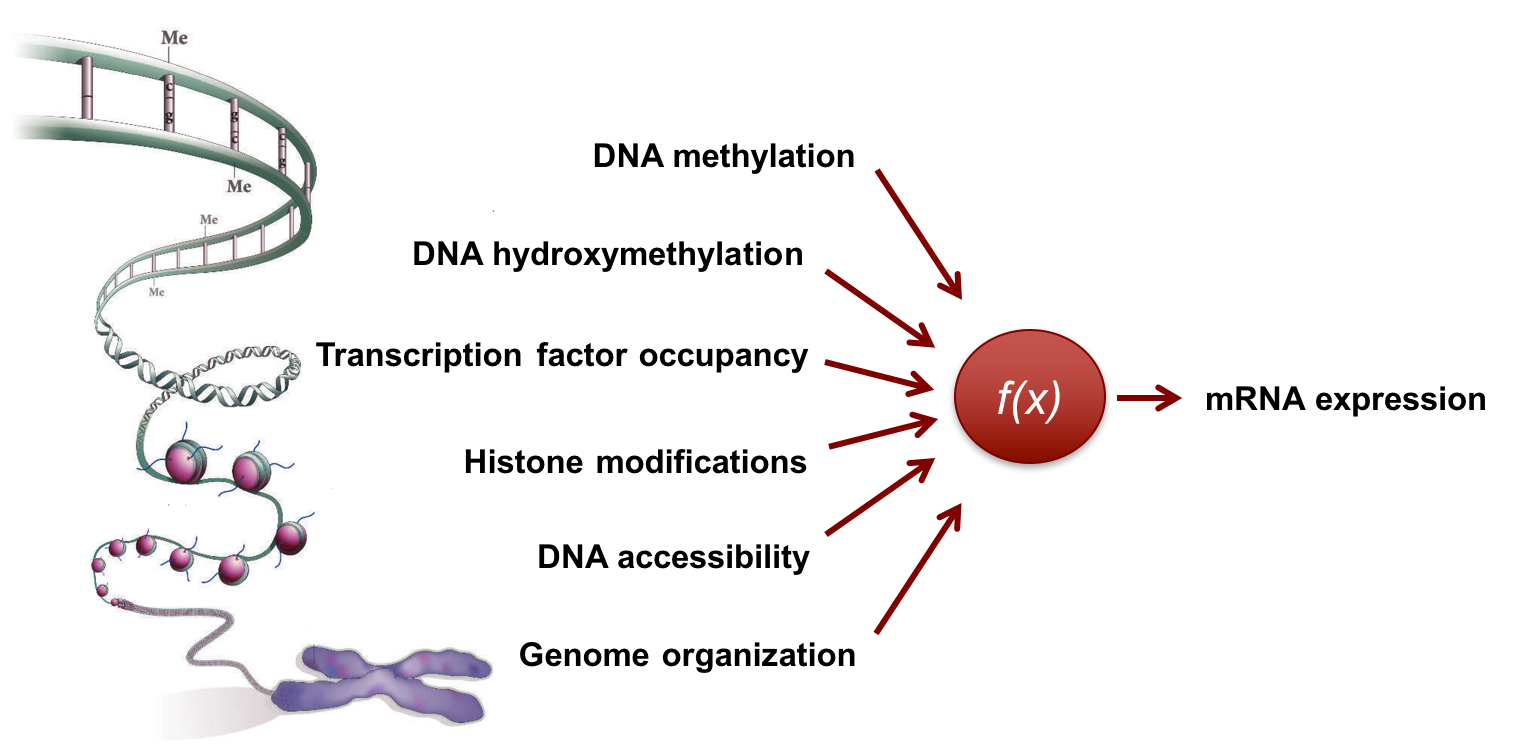 |
Understanding how the genome/epigenome regulates gene expression and cellular phenotypes is a fundamental question in biology. The advent of next-general sequencing technologies has heralded a new era where correlations between the genome/epigenome and the transcriptome can be established on a genome-wide scale. However, as these measurements are made starting from a large population of cells or a complex tissue, they provide only an ensemble average description of the system. |
|
|
To overcome these challenges and to directly associate variations in the genome/epigenome to gene expression heterogeneity, we develop technologies that enable simultaneous genome-wide measurements of the epigenome and transcriptome from the same cell. We recently developed the first integrated method to sequence both the genome and transcriptome from the same cell to study how copy number variations in cancer influence cell-to-cell heterogeneity in gene expression. Similarly, we have developed technologies to sequence epigenetic marks such as DNA methylation (5mC) and DNA hydroxymethylation (5hmC) in single cells as well as developed tools to quantify the organization of the genome in single cells. These measurements have revealed dramatic cell-to-cell variability in the epigenome and we are currently developing integrated single-cell technologies to study how this variability in the epigenome regulates gene expression heterogeneity and cellular outcomes. |
||
Cellular lineage reconstruction using single-cell epigenomics
|
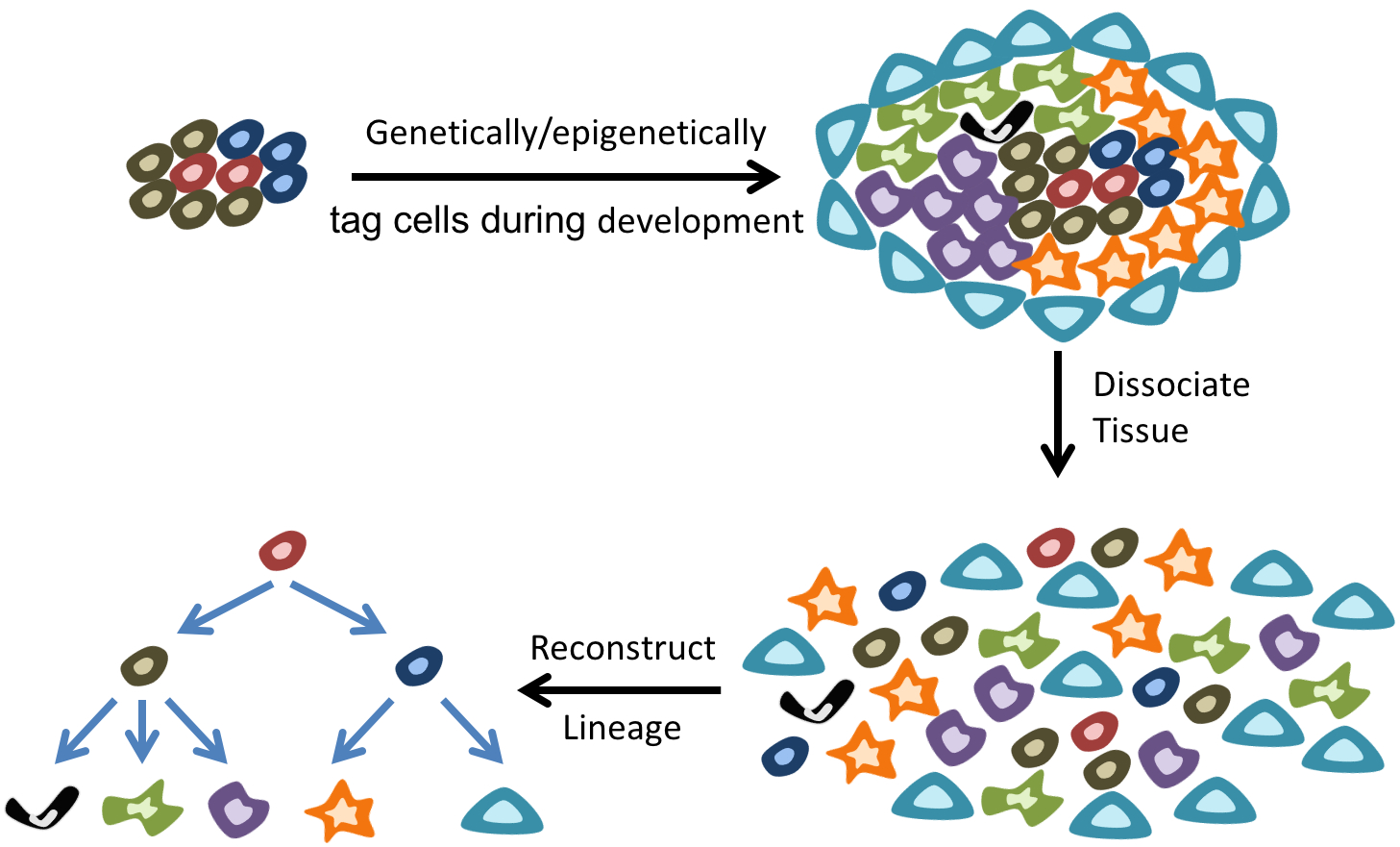
While single-cell mRNA sequencing is providing an unprecedented view of cell types within a tissue, an important step towards better characterization of the dynamics of tissue development and homeostasis is to map the evolutionary lineage tree connecting cells. Surprisingly, our understanding of cellular lineage trees for many tissues in multicellular organisms, except simple organisms such as C. elegans, is extremely limited. Traditionally, tracing and reconstructing lineage trees has relied on the use of complex transgenic animal models or other exogenous agents (such as viruses or dyes). In our group, we are developing novel single-cell epigenome sequencing technologies to endogenously reconstruct cellular lineages during early mouse development. These technologies will provide a deeper understanding of the factors regulating cell fate decisions and their role in dynamically regulating tissue composition. |
 |